Utiliser notre service RMN qui fournit la RMN 1H et de nombreuses autres techniques RMN.
Il existe trois isotopes de l’hydrogène utilisés en spectroscopie RMN : 1Hydrogène, 2Deutérium et 3Tritium. Chaque isotope résonne à une fréquence très différente ; par exemple, si 1H résonne à 400 MHz, alors 2H résonne à 61,402 MHz. Un seul isotope est observé à la fois car le spectromètre émet et reçoit sur une gamme de fréquences très limitée. Les plages de déplacement chimique des trois noyaux sont pratiquement identiques et peuvent être utilisées pour une analyse préliminaire, mais la similitude s’arrête là. 3Le tritium n’est pas couramment mesuré par RMN car il est radioactif.
Chaque type de signal a une plage de déplacement chimique caractéristique (fig. 1) qui peut être utilisée pour une affectation initiale.
Fig. 1. Gammes de déplacement chimique des protons en fonction de leur environnement chimique
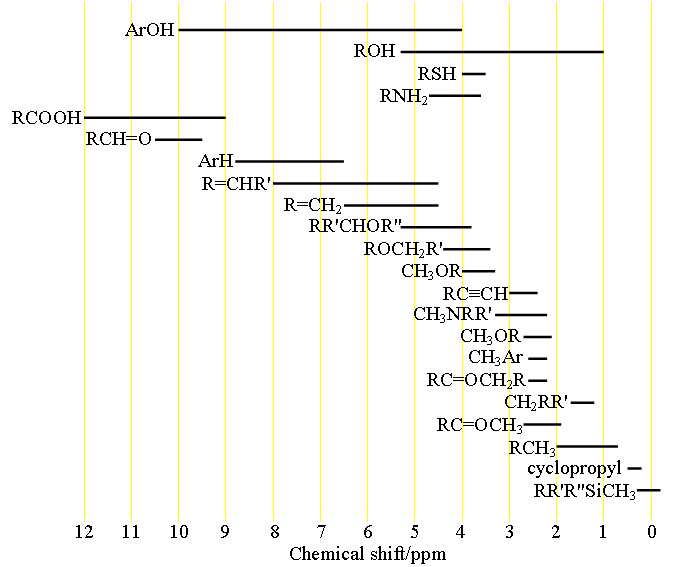
Choisissez la structure qui représente le mieux l’hydrogène en question. R = alkyle ou H, Ar = aryle.
1Hydrogène (proton) RMN
L’expérience 1D 1H (proton) RMN est l’expérience RMN la plus courante. Le proton (noyau 1Hydrogène) est le noyau le plus sensible (hormis le tritium) et donne généralement des signaux nets. Même si sa plage de déplacement chimique est étroite, ses signaux nets rendent la RMN du proton très utile. Notre service RMN propose la RMN du proton ainsi que de nombreuses autres techniques de RMN.
Une analyse typique d’un spectre RMN 1H peut se dérouler comme suit :
Le nombre de protons de chaque type dans le spectre d’un échantillon pur peut être obtenu directement à partir des intégrales de chaque multiplet. Ceci n’est vrai que si les multiplets sont bien séparés et ne recouvrent pas les signaux du solvant ou de l’eau résiduelle et à condition que la molécule ne subisse pas un échange conformationnel lent. Un spectre RMN de routine donne des intégrales avec une précision de +/-10%. Des précisions de +/-1% peuvent être obtenues en augmentant le délai de relaxation à cinq fois le temps de relaxation longitudinal (T1) des signaux d’intérêt. Lorsque les multiplets se chevauchent, on peut utiliser l’intégrale totale de la région spectrale.
À partir du tableau des déplacements chimiques des protons, on obtient des informations sur chaque type de proton et on peut effectuer une affectation préliminaire.
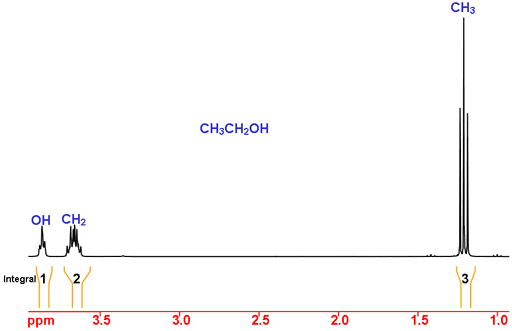
Prenons l’exemple de l’éthanol (figure 2). En utilisant les déplacements chimiques, le pic à 1,2 ppm est dans la gamme attendue pour CH3 (méthyle) et à 3,7 et 3,9 ppm sont compatibles avec CH2 (méthylène). Le déplacement chimique de OH est très dépendant du solvant et des autres conditions expérimentales, il ne peut donc pas être attribué par le déplacement chimique seul.
En utilisant l’intégration, nous nous attendons à ce que CH3 ait une intégrale de trois, CH2 une intégrale de deux et OH une intégrale de un. C’est le cas et l’affectation est donc complète.
Fig. 2. Spectre RMN 1H de l’éthanol dans le CDCl3
Pour d’autres molécules, cela n’est pas suffisant et la structure des multiplets est nécessaire pour compléter l’affectation. Les multiplets (fig. 3) proviennent des couplages spin-spin qui sont transmis par les liaisons chimiques et donnent des informations sur l’environnement moléculaire immédiat. Dans le cas de CH3 et OH, ils sont divisés par les deux protons voisins du CH2 pour donner une structure de triplet appelée AX2. (Cliquez ici pour voir une liste des motifs de division homonucléaires courants et une description du couplage hétéronucléaire). Le CH2 est divisé par l’unique proton OH et les trois protons CH3 pour former le motif AX3Y. (Cliquez ici pour d’autres exemples d’affectation spectrale, d’affectation 2D du 12,14-ditbutylbenzochrysène et d’affectation 2D de l’acétate de cholestéryle.)
Fig. 3. Structures de multiplets à partir du spectre RMN 1H de l’éthanol dans le CDCl3
Ayant déterminé la multiplicité, on peut mesurer les constantes de couplage. Celles-ci sont mesurées en Hz (et non en ppm), car elles sont indépendantes du champ. Si vous trouvez que deux multiplets (et seulement deux) contiennent la même constante de couplage, alors vous savez qu’ils proviennent de protons proches. La constante de couplage donne une indication de la distance entre les protons. En général, 10 à 18 Hz signifie que les multiplets sont trans à deux ou trois liaisons vers une double liaison C=C. Les constantes de couplage entre 1 et 10 Hz indiquent trois liaisons ou plus si elles sont délocalisées. Moins de 1 Hz signifie généralement quatre liaisons ou plus.
En plus des couplages homonucléaires, les multiplets peuvent être divisés par d’autres noyaux tels que 19Fluor ou 31Phosphore. (Si de tels couplages hétéronucléaires ne sont pas souhaitables, ils peuvent être découplés. La meilleure séquence d’impulsions dans un tel cas est celle du découplage par gate inverse.)
Propriétés de 1H
(Cliquez ici pour une explication)
| Propriété | Valeur |
|---|---|
| Spin | ½ |
| Abondance naturelle | 99.9845% | Etendue du décalage chimique | 13 ppm, de -1 à 12 | Rapport de fréquence (Ξ) | 100.000000% | Composé de référence | TMS < 1% dans CDCl3 = 0 ppm |
| Largeur de ligne de la référence | 0.08 Hz | T1 de la référence | 14 s | Réceptivité par rapport à 1H à l’abondance naturelle | 1.000 |
| Réceptivité relative à 1H lorsqu’il est enrichi | 1,000 | Réceptivité relative. au 13C à l’abondance naturelle | 5870 | Réceptivité rel. au 13C lorsqu’il est enrichi | 5871 |
RMN du 2Deutérium
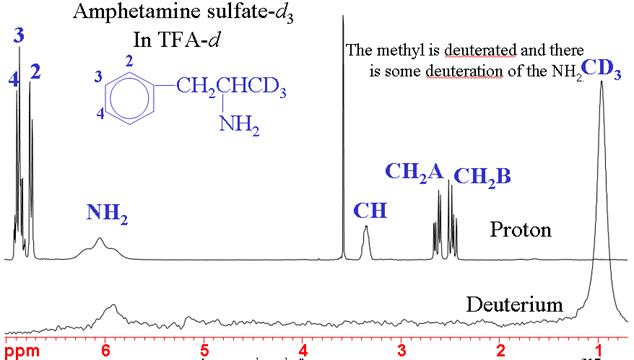
La RMN du 2Deutérium (hydrogène lourd) est généralement utilisée pour le verrouillage de la fréquence du champ. A l’abondance naturelle, il a une très faible sensibilité mais lorsqu’il est enrichi, il est de sensibilité moyenne. Le deutérium donne habituellement des signaux larges dont la largeur de ligne varie typiquement entre quelques hertz et quelques kilohertz. Le spectre présente la même gamme étroite de déplacements chimiques que pour le 1H, mais sa faible résolution et sa plus faible sensibilité en font une mauvaise alternative. Les couplages deutérium-deutérium sont environ 40 fois plus petits que les couplages proton-proton et ne sont donc pas observés. Cependant, dans les molécules partiellement deutérées, de petits couplages proton-deutérium peuvent être observés. La principale utilisation des spectres de deutérium est de déterminer l’efficacité de la deutération chimique (fig. 4).
Fig. 4. Spectres RMN 1H et 2H du sulfate d’amphétamine-d3 montrant la réussite de la deutération spécifique du méthyle
Propriétés de 2H
(Cliquez ici pour une explication)
| Propriété | Valeur |
|---|---|
| Spin | 1 | Abondissement naturel | 0.0155% | Etendue du décalage chimique | 13 ppm, de -1 à 12 | Rapport de fréquence (Ξ) | 15.350609% | Composé de référence | TMS-d12 pur = 0 ppm | Largeur de ligne de la référence | 1.7 Hz | T1 de la référence | 1 s | Réceptivité relative à 1H en abondance naturelle | 1,50 × 10-6 |
| Réceptivité relative à 1H lorsqu’il est enrichi | 9.65 × 10-3 | Réceptivité relative au 13C à l’abondance naturelle | 8,78 × 10-3 |
| Réceptivité relative au 13C lorsqu’il est enrichi | 56.7 |
| Paramètre de largeur de ligne | 0,41 fm4 |
RMN du 3Tritium
Le 3T est le seul noyau plus sensible que le proton (1H). Étant un isotope de spin-½ de l’hydrogène, les spectres des composés entièrement tritiés ressemblent à ceux de 1H avec effectivement les mêmes déplacements chimiques mais avec une sensibilité, une dispersion et des constantes de couplage légèrement plus élevées. Cependant, le 3T est très radioactif, de sorte que la plupart des études RMN sont réalisées avec des composés partiellement et spécifiquement marqués.
Notre laboratoire ne dispose pas de l’équipement nécessaire pour manipuler le tritium ou acquérir des spectres RMN du tritium. Cependant, nous sommes prêts à discuter de la logistique s’il y a une demande pour ce service.
Propriétés de 3H
(Cliquez ici pour une explication)
| Propriété | Valeur |
|---|---|
| Spin | ½ |
| Abondance naturelle | 0.0000000000000003% | Etendue du décalage chimique | 13 ppm, de -1 à 12 | Rapport de fréquence (Ξ) | 106.663974% | Composé de référence | TMS-t1 < 1% dans CDCl3 = 0 ppm | Largeur de ligne de la référence | ~0.1 Hz | T1 de référence | ~20 s | Réceptivité rel. à 1H à l’abondance naturelle | 4 × 10-18 |
| Réceptivité rel. à 1H lorsqu’il est enrichi | 1,21 |
| Receptivité rel. à 13C à l’abondance naturelle | 2 × 10-14 |
| Receptivité rel. au 13C lorsqu’il est enrichi | 7103 |
Note de sécurité:
Certaines des matières mentionnées ici sont très dangereuses. Demandez conseil à un chimiste qualifié avant de les manipuler. Les chimistes qualifiés doivent consulter la littérature de sécurité pertinente avant de manipuler ou de donner des conseils sur des substances non familières. Les solvants de RMN sont toxiques et la plupart sont inflammables. En particulier, le TMS est toxique, volatile et inflammable : portez des gants de protection et travaillez sous une hotte. Tous les composés deutérés sont toxiques. Le tritium est radioactif : assurez-vous d’avoir l’expertise et l’équipement nécessaires avant de le manipuler.