Utilice nuestro servicio de RMN que proporciona RMN de 1H y muchas otras técnicas de RMN.
Hay tres isótopos de hidrógeno utilizados en la espectroscopia de RMN: 1Hidrógeno, 2Deuterio y 3Tritio. Cada isótopo resuena a una frecuencia muy diferente, por ejemplo, si el 1H resuena a 400 MHz, el 2H resuena a 61,402 MHz. Sólo se observa un isótopo a la vez porque el espectrómetro transmite y recibe en un rango de frecuencia muy limitado. Los rangos de desplazamiento químico para los tres núcleos son prácticamente idénticos y pueden utilizarse para un análisis preliminar, pero ahí termina la similitud. El tritio no se suele medir por RMN porque es radiactivo.
Cada tipo de señal tiene un rango de desplazamiento químico característico (fig. 1) que puede utilizarse para la asignación inicial.
Fig. 1. Rangos de desplazamiento químico de los protones según su entorno químico
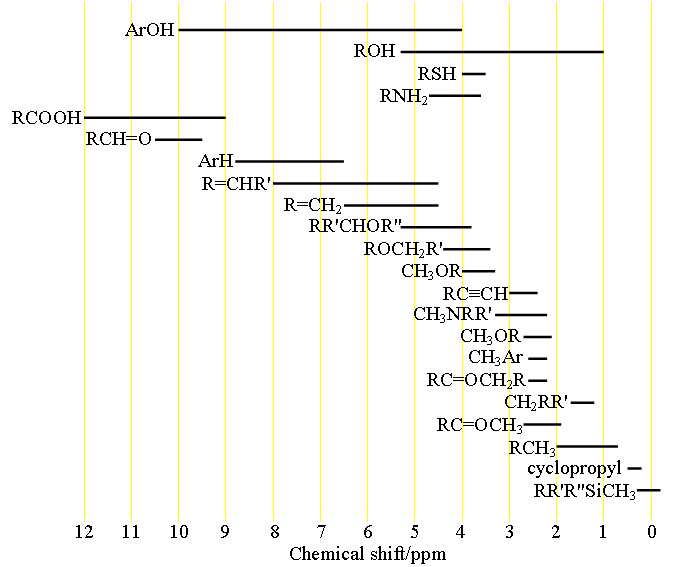
Elija la estructura que más represente al hidrógeno en cuestión. R = alquilo o H, Ar = arilo.
RMN de 1H (protón)
El experimento de RMN de 1D 1H (protón) es el más común. El protón (núcleo de 1Hdrógeno) es el núcleo más sensible (aparte del tritio) y suele producir señales nítidas. Aunque su rango de desplazamiento químico es estrecho, sus señales nítidas hacen que la RMN de protones sea muy útil. Nuestro servicio de RMN ofrece la RMN de protones junto con muchas otras técnicas de RMN.
Un análisis típico de un espectro de RMN de 1H puede proceder como sigue:
El número de protones de cada tipo en el espectro de una muestra pura puede obtenerse directamente de las integrales de cada multiplete. Esto sólo es cierto si los multipletes están bien separados y no se solapan con las señales del disolvente o del agua residual y siempre que la molécula no esté sufriendo un intercambio conformacional lento. Un espectro de RMN de rutina produce integrales con una precisión de +/-10%. Pueden alcanzarse precisiones de +/-1% aumentando el retardo de relajación a cinco veces el tiempo de relajación longitudinal (T1) de las señales de interés. Cuando los multipletes se superponen, se puede utilizar la integral total de la región espectral.
A partir de la tabla de los desplazamientos químicos de los protones se obtiene información sobre cada tipo de protón y se puede realizar una asignación preliminar.
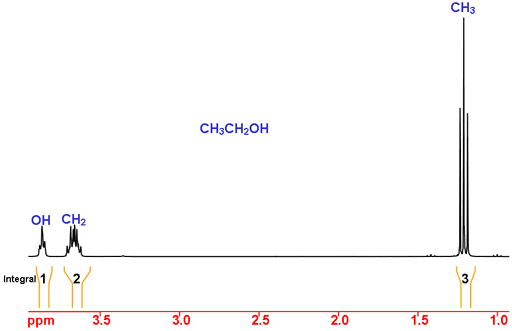
Considere el etanol como ejemplo (Fig. 2). Utilizando los desplazamientos químicos, el pico a 1,2 ppm está en el rango esperado para el CH3 (metilo) y a 3,7 y 3,9 ppm son compatibles con el CH2 (metileno). El desplazamiento químico del OH depende en gran medida del disolvente y de otras condiciones experimentales, por lo que no puede asignarse únicamente mediante el desplazamiento químico.
Utilizando la integración, esperamos que el CH3 tenga una integral de tres, el CH2 una integral de dos y el OH una integral de uno. Este es el caso y por tanto la asignación está completa.
Fig. 2. Espectro de RMN de 1H del etanol en CDCl3
Para otras moléculas esto no es suficiente y se necesita la estructura de los multipletes para completar la asignación. Los multipletes (fig. 3) surgen de los acoplamientos espín-espín que se transmiten a través de los enlaces químicos y proporcionan información sobre el entorno molecular inmediato. En el caso del CH3 y el OH, son divididos por los dos protones vecinos del CH2 para dar lugar a un patrón de tripletes llamado AX2. (Haga clic aquí para ver una lista de patrones de desdoblamiento homonucleares comunes y una descripción del acoplamiento heteronuclear). El CH2 es dividido por el único protón OH y los tres protones CH3 para formar el patrón AX3Y. (Haga clic aquí para ver más ejemplos de asignación espectral, asignación 2D del 12,14-ditbutilbenzoquisteno y asignación 2D del acetato de colesterol.)
Figura 3. Estructuras de multipletes del espectro de RMN de 1H del etanol en CDCl3
Habiendo determinado la multiplicidad, se pueden medir las constantes de acoplamiento. Estas se miden en Hz (no en ppm), ya que son independientes del campo. Si se encuentra que dos (y sólo dos) multipletes contienen la misma constante de acoplamiento, se sabe que provienen de protones cercanos. La constante de acoplamiento da una indicación de la distancia entre los protones. En general, de 10 a 18 Hz significa que hay dos o tres enlaces trans a un doble enlace C=C. Las constantes de acoplamiento entre 1 y 10 Hz indican tres enlaces o más enlaces si están deslocalizados. Menos de 1 Hz suele significar cuatro o más enlaces.
Además de los acoplamientos homonucleares, los multipletes pueden ser divididos por otros núcleos como el 19Fluoro o el 31Fósforo. (Si estos acoplamientos heteronucleares no son deseables, pueden desacoplarse. La mejor secuencia de pulsos en tal caso es la del desacoplamiento gated inverso.)
Propiedades del 1H
(Pulse aquí para ver la explicación)
| Propiedad | Valor |
|---|---|
| Giro | ½ |
| Abundancia natural | 99.9845% |
| Gama de desplazamiento químico | 13 ppm, de -1 a 12 |
| Relación de frecuencia (Ξ) | 100.000000% |
| Compuesto de referencia | TMS < 1% en CDCl3 = 0 ppm |
| Ancho de línea de referencia | 0.08 Hz |
| T1 de la referencia | 14 s |
| Receptividad relativa a 1H en abundancia natural | 1.000 | Receptividad rel. a 1H cuando está enriquecido | 1.000 | Receptividad rel. al 13C en abundancia natural | 5870 | Receptividad rel. al 13C cuando está enriquecido | 5871 |
RMN de 2Deuterio
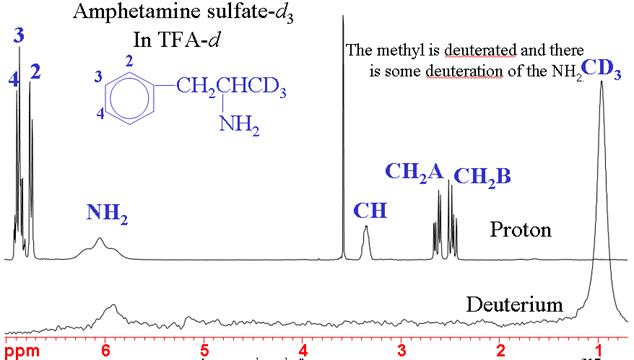
La RMN de 2Deuterio (hidrógeno pesado) se utiliza normalmente para el bloqueo de la frecuencia de campo. En su abundancia natural tiene una sensibilidad muy baja, pero cuando está enriquecido es de sensibilidad media. El deuterio suele producir señales amplias cuyo ancho de línea suele variar entre unos pocos hercios y unos pocos kilohercios. El espectro tiene el mismo rango de desplazamiento químico estrecho que el del 1H, pero su baja resolución y su menor sensibilidad lo convierten en una mala alternativa. Los acoplamientos deuterio-deuterio son unas 40 veces menores que los acoplamientos protón-protón y, por tanto, no se observan. Sin embargo, en las moléculas parcialmente deuteradas pueden observarse pequeños acoplamientos protón-deuterio. El principal uso de los espectros de deuterio es para determinar la eficacia de la deuteración química (fig. 4).
Fig. 4. Espectros de RMN de 1H y 2H del sulfato de anfetamina-d3 que muestran una deuteración específica exitosa del metilo
Propiedades del 2H
(Pulse aquí para ver la explicación)
| Propiedad | Valor |
|---|---|
| Giro | 1 |
| Abundancia natural | 0.0155% |
| Gama de desplazamiento químico | 13 ppm, de -1 a 12 |
| Relación de frecuencia (Ξ) | 15.350609% | Compuesto de referencia | TMS-d12 neat = 0 ppm | Ancho de línea de referencia | 1.7 Hz |
| T1 de referencia | 1 s | Receptividad relativa a 1H en abundancia natural | 1,50 × 10-6 |
| Receptividad relativa a 1H cuando está enriquecido | 9.65 × 10-3 | Receptividad relativa al 13C en abundancia natural | 8,78 × 10-3 | Receptividad relativa al 13C cuando está enriquecido | 56.7 |
| Parámetro de anchura de línea | 0,41 fm4 |
La RMN del 3T es el único núcleo más sensible que el protón (1H). Al tratarse de un isótopo de hidrógeno de spin-½, los espectros de los compuestos totalmente tritiados tienen un aspecto similar al del 1H, con efectivamente los mismos desplazamientos químicos pero con una sensibilidad, dispersión y constantes de acoplamiento ligeramente superiores. Sin embargo, el 3T es muy radiactivo, por lo que la mayoría de los estudios de RMN se llevan a cabo con compuestos parcial y específicamente etiquetados.
Nuestro laboratorio no cuenta con el equipo necesario para manejar tritio o adquirir espectros de RMN de tritio. Sin embargo, estamos dispuestos a discutir la logística si hay una demanda de este servicio.
Propiedades del 3H
(Pulse aquí para ver la explicación)
| Propiedad | Valor |
|---|---|
| Giro | ½ |
| Abundancia natural | 0.0000000000000003% | Gama de desplazamiento químico | 13 ppm, de -1 a 12 |
| Relación de frecuencias (Ξ) | 106.663974% | Compuesto de referencia | TMS-t1 < 1% en CDCl3 = 0 ppm | Ancho de línea de referencia | ~0.1 Hz |
| T1 de referencia | ~20 s |
| Receptividad rel. a 1H en abundancia natural | 4 × 10-18 | Receptividad rel. al 1H cuando está enriquecido | 1,21 | Receptividad rel. al 13C en abundancia natural | 2 × 10-14 | Receptividad rel. al 13C cuando está enriquecido | 7103 |
Nota de seguridad:
Algunos de los materiales mencionados aquí son muy peligrosos. Pida consejo a un químico cualificado antes de manipularlos. Los químicos cualificados deben consultar la bibliografía de seguridad correspondiente antes de manipular o aconsejar sobre sustancias desconocidas. Los disolventes de RMN son tóxicos y la mayoría son inflamables. En concreto, el TMS es tóxico, volátil e inflamable: utilice guantes de protección y trabaje en una campana. Todos los compuestos deuterados son tóxicos. El tritio es radiactivo: asegúrese de tener los conocimientos y el equipo necesarios antes de manipularlo.